
Press-room / news / Science news /
Origin, Evolution and Diversity of φ29-like Phages
The application of the definition of taxonomic species to the description of evolutionary history of biological objects with chimeric genomes is a difficult task. The limited period of the existence of species and the lability of viral genomes can also make attempts to reconstruct the evolutionary history almost pointless. The processes of genetic exchange seem to be especially pronounced in temperate phages, but genetic rearrangements between evolutionarily close groups of phages can also affect φ29-like phages. However, we can try to plot the evolutionary traits of genes encoding separate essential proteins and their stable complexes.

The group of φ29-like phages possesses pronounced unique features in packaging and replication, which are interrelated processes. Phages of this group have a similar architecture of their genomes and similar virion proteins (e.g., capsid and portal proteins). Structural interactions between morphogenesis proteins drives the co-evolution of these proteins and may be one of the reasons for the stability of φ29-like genomes. HMM and BLAST searches using the sequences of the major capsid protein and terminase carried out in this study allowed us to identify relative phages. The results indicate that the set of main virion proteins (p6-p11) is the hallmark feature of φ29-like group, though their sequences may significantly differ from the sequences of other phage proteins. The proteins comprising replication complex and terminase are also characteristic for these phages.
The present research indicates that the evolution of φ29-like viruses was accompanied by genome rearrangements, gene duplication and loss, and the acquisition of domains of apparently cellular origin. The latter applies even to such a conserved protein as the major capsid protein. Notably, a comparison of the structures of the φ29 major capsid protein (excluding the Ig-like domain presumably acquired by horizontal transfer) showed that the φ29 MCP is more similar to its counterpart from Autographiviridae phages than to its counterparts from other groups of Caudoviricetes phages. This could be an argument for the evolutionary relatedness of φ29-like and T7-like phages. The unlikeness of amino acid sequences of MCP of φ29-like phages to sequences of MCPs of all other Duplodnaviria viruses (except phage KSY1), including T7-like phages, however, which has been confirmed by HMM searches, is a strong indication of the ancient divergence of the φ29-like phage group.
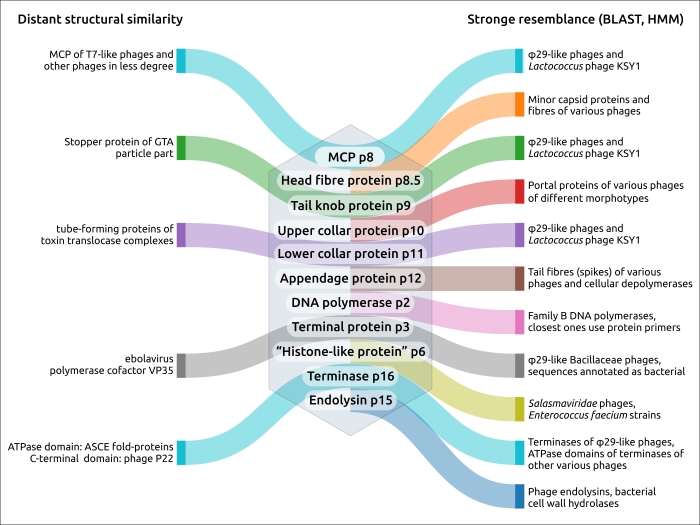
Figure 1. Diagram indicating the close and distant relatedness of φ29 proteins, based on the conducted analyses and earlier-published data.
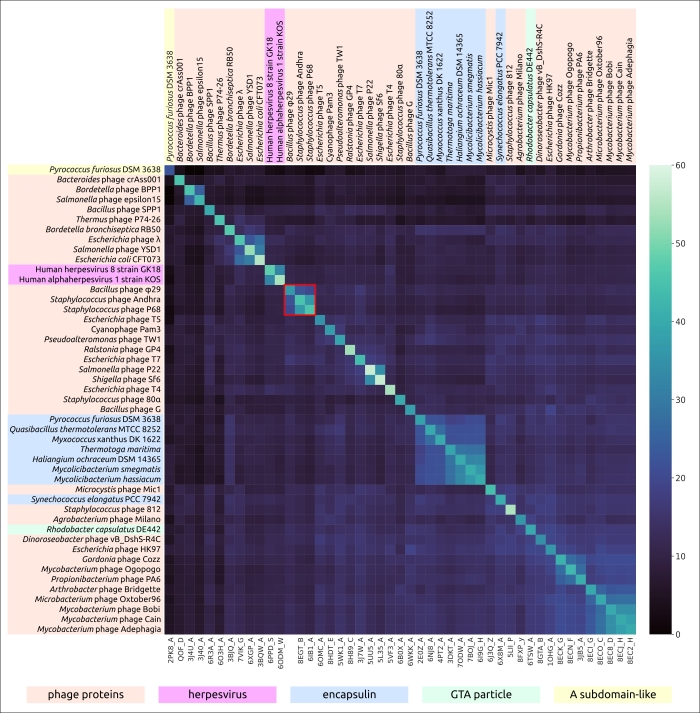
Figure 2. Heatmap based on the pairwise structural similarity of viral capsid proteins and encapsulin major shell proteins, as measured by DALI Z-scores. The cluster of φ29-related phages is outlined in red.
Unexpectedly, HMM searches revealed that a peculiar phage, Lactococcus phage KSY1 (Chopinvirus KSY1), which is very much not like φ29 in genome size and content, has φ29-like terminase and the virion proteins, including homologues of φ29 MCP and non-RBP tail proteins. Perhaps φ29-like phages are just a part of an even larger clade of evolutionarily related bacteriophages. Seemingly, Lactococcus phage KSY1 acquired genes of adsorption apparatus from other phages infecting Lactococcus or related bacteria, but the source of the genes involved in transcription and replication requires additional research. However, a sequence search has clearly indicated the relatedness of Chopinvirus, Autographiviridae and Schitoviridae RNA polymerases, and phylogenetic analysis has indicated that the RNAPs of all three taxa belong to different clades. It will be interesting to see if the Lactococcus phage KSY1 can help find the link between the T7- and φ29-like viruses. In spite of having transcription apparatus resembling T7-like phages, the genome of phage KSY1 contains neither T7-like DNA polymerase nor φ29-like DNAP. According to the results of phylogenetic analysis, the latter is related to the DNAP of tectiviruses.
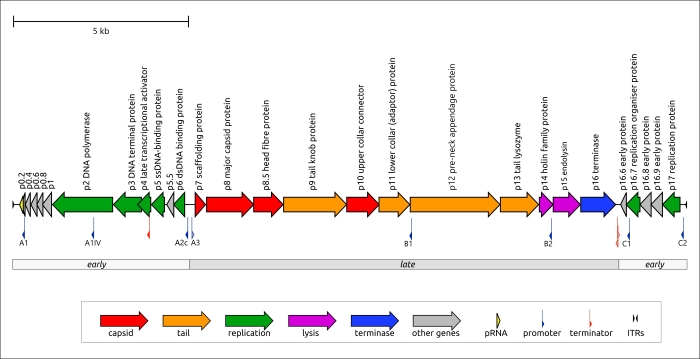
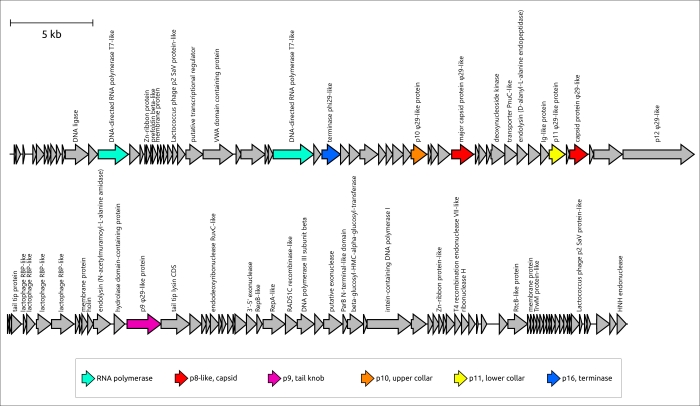
Figure 3. Genetic map of Bacillus phage φ29 and Lactococcus phage KSY1. Arrows indicate the direction of transcription. The scale bar indicates the length of the nucleotide sequence. Gene functions are shown in labels and the legend.
Although some evolutionary relatives of long-tailed phages are known, the details of the evolution of short-tailed phages are less understood. Actually, some domains of phage structural proteins, especially those participating in adsorption and cell-wall degradation, can have a resemblance with proteins or protein domains of cellular origin, as well as with proteins of viral origin, even those encoded in genomes of phages belonging to distant viral groups, but these cases are apparently the result of horizontal transfer. From this point of view, the results of the current study, which indicate the resemblance of the φ29 tail tube formed by lower collar protein p11 and tail tubes of toxin translocase complexes of Gram-positive bacteria, could offer a partial explanation of the origin of the tail of φ29-like phages. A search for similar structures using non-RBP tail proteins of φ29, excluding upper collar (portal) protein, which is common for all tailed phages and herpesviruses, and which could be considered to be part of the tail head (capsid), did not find similar proteins among podoviruses belonging to well-studied groups of T7-like phages (family Autographiviridae) and N4-like phages (family Schitoviridae). A previous publication indicates that the amino-terminal domain of the tail knob proteins of a φ29-like phage C1 infecting Streptococcus, and apparently of φ29 itself, has a fold found in tube-forming proteins of sipho- and myoviruses phages, indicating evolutionary ties between long-tailed and short-tailed phages. A similar fold was found in phage and herpesvirus proteases, as well as in the tail stopper protein of the GTA of Rhodobacter capsulatus, structurally resembling p11 according to DALI. However, this fold seems to be missing the protein structures of another podoviral tail tube, namely of phage T7 (see, for example, PDB #6R21). Perhaps the morphological classification of podoviruses needs some clarification, since their tails may have different origin and architecture.
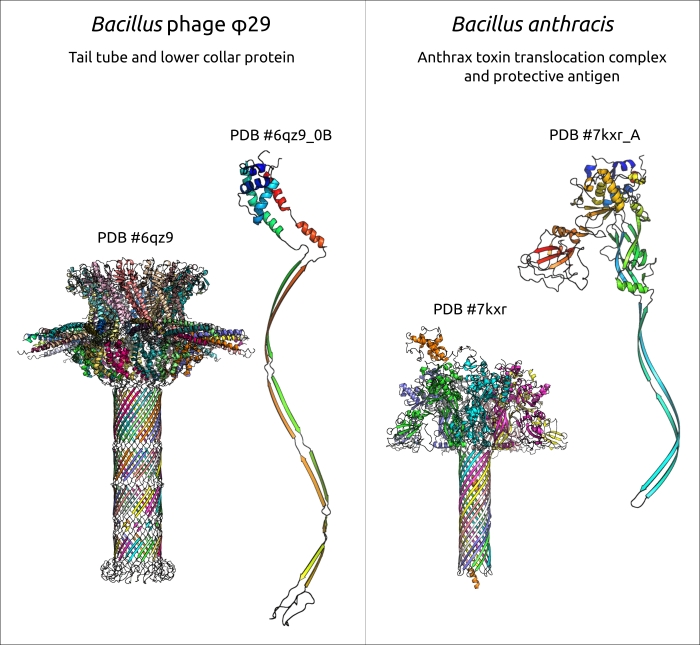
Figure 4. The tail tube of phage φ29 and lower collar protein p11, and anthrax toxin translocation complex and protective antigen protein from Bacillus anthracis. In ternary complexes, different chains have a different colour. Separate monomers are shown using rainbow colouring, with a colour gradient where the N-terminal end is blue and the C-terminus is red.
The evolution of φ29-like phages may involve both coevolution with the host and host switching, based on an analysis and comparisons of phylogenetic trees relating to phage and bacterial sequences. Members of φ29-like groups of phages are able to infect a wide range of hosts, and the analysis of metagenomic sequences indicated that, in addition to bacteria of the Bacillales, Clostridia, Enterobacterales and Pseudomonadales orders, these phages might be able to infect representatives of the Bacteroidales order. Perhaps the phage Bacteriophage sp. isolate 2692_55609, representing cluster c30, is the earliest diverged φ29-like phage (among the analysed representatives) which infects Gram-negative bacteria. A phylogenetic analysis of MCP and DNAP indicates that this phage is related to Amedibacillus phage AD70P2. Amedibacillus bacteria are phylogenetically distant from Gram-negative bacteria. The origin and evolution of φ29-like phages infecting Gram-negative bacteria is probably linked to host switching followed by modification of some proteins. Particularly, Gram-negative-specific phages have shortened lower collar (tail tube) protein, which may be due to the thinner cell wall of the bacterial host. Also, the recent publication on Acinetobacter phage Phanie and the results of this work suggest the distinct composition of the tail knob in phages infecting Gram-negative bacteria. The tail knob of analysed φ29-like phage infecting Gram-negative bacteria appears to be a hexamer, each unit of which consists of two proteins. One of them is located closer to the phage head and corresponds to N-terminal part of φ29 tail knob protein p9, and the second distal protein is analogous to the carboxy-terminal part of φ29 p9. This split of the gene encoding the tail knob protein probably occurred in the ancestor of φ29-like phages infecting Gram-negative bacteria due to genetic exchange, and resulted in the insertion of the fragment encoding Ig-like domains. It has been shown that Ig-like domains can participate in the interaction with carbohydrates on the surface of the bacterial cell and participate in the formation of phage tail appendages and sheaths. The results of structural predictions suggest that Ig-like domains are located lateral to the tail tube. Particularly, the AF model of the protein complex of a tail knob of Pectobacterium phage DU_PP_III, predicted with a decent quality (ipTM score 0.69, pTM score 0.70), depicts the Ig-like domains down to the phage head as resembling short tail fibres, though with architecture fundamentally different from common trimeric architecture. Acinetobacter phage Phanie contains six such domains in gp11. They resemble the Ig-like non-catalytic domain of L,D-transpeptidase from Mycobacterium tuberculosis; hypothetically, these domains are able to exhibit carbohydrate-binding properties. The formation of chimeric virions of phage Phanie and helper myovirus Acinetobacter phage Aci01-1 was experimentally demonstrated. Based on TEM images, myoviral tail tube is attached distally to the tail of phage Phanie. In a previous work, it was suggested that the additional Ig-like domains of a myoviral tail sheath protein can facilitate increasing the stability of the sheath due to protein–protein interactions involving additional domains. It is probable that these domains play an important role in the formation of chimeric virions.
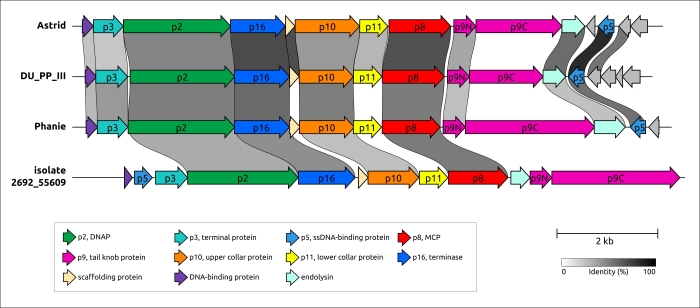
Figure 5. Genetic map of phages belonging to clusters 23 and 30. Phages’ names are as follows: Astrid, Salmonella phages Astrid; DU_PP_III, Pectobacterium phage DU_PP_III; Phanie, Acinetobacter phage Phanie; isolate 2692_55609, Bacteriophage sp. isolate 2692_55609. Annotations based on the results of HMM searches, AF predictions and an earlier-published genomic analysis of phage Phanie are shown in labels. Arrows indicate the direction of transcription. The scale bar indicates the level of identity between genes according to a black–white gradient, the legend for which is shown at the bottom of the figure. The number of clusters and the name of sequences are shown on the left. Genes encoding proteins similar to φ29 (p2, p5, p6, p8–p11, p16) and other proteins are coloured according to the legend.
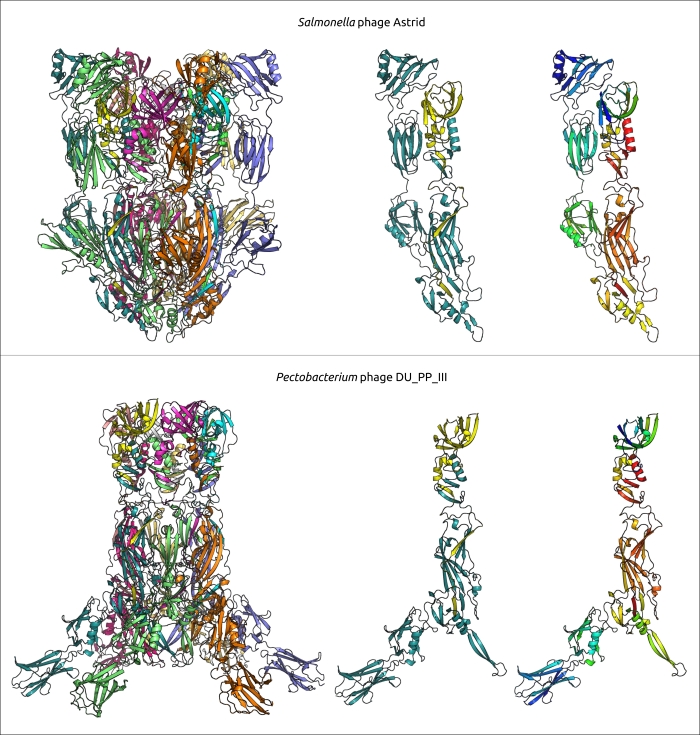
Figure 6. A ribbon diagram of the protein complexes, modelled by AlphaFold3, supposedly corresponding to tail knobs of Salmonella phages Astrid and Pectobacterium phage DU_PP_III. Left depictions show the full-sized model comprising 12 monomers (each monomer has a different colour). The central depiction shows two closest different monomers, the protein similar to the N-terminal part of the φ29 tail knob protein is coloured yellow, and the protein similar to the C-terminal part is coloured cyan. The right-side depictions show the same monomers as the central one using a rainbow colour gradient, where the N-terminal end of each monomer is blue and the C-terminus is red.
Summarising the aforesaid, the group of φ29-like phage clearly differs from other phage groups. It has much in common, though it has variations in genomes, bacterial hosts and infection mechanisms. A search for φ29-like and related viruses conducted for the present study could not find many members of this group, since it employed the HMM-based method on a limited database. The search did, however, identify more than 2000 sequences, indicating the great abundance of these viruses. The prevalence of φ29-related viruses, the complex of unique features, including the characteristic morphology and replication processes, and their distinctive origin, including the early divergence of conserved phage proteins, suggest the possibility of delineating this group as a separate taxon. Perhaps it should be ranked at the level of an order or higher.
The results are published in the International Journal of Molecular Sciences.
october 11, 2024